|
Chapter 4
The adrenal glands in gynaecology
The adrenal glands are part of the neuroendocrine hypothalamo-pituitary-adrenal (HPA) axis. They are essential for our survival and are involved in orchestrating almost all body functions, especially cortisol which has receptors in all tissues in the body. This importance is reflected by the fact that the HPA axis is functional by the 6th week of intrauterine fetal life. Furthermore, each adrenal gland is richly supplied by 3 arteries despite their small size. During fetal life, each gland is made of an inner medulla and an outer cortex which is made of a small outer adult zone and an inner and much larger functional fetal zone. This fetal zone shrinks after birth and is replaced by the zona reticularis as from the 4th or 5th year of life. The outer adult zone, on the other hand, increases in size with time. The inner medulla is derived from the neuroectoderm, and the outer cortex is formed from mesoderm. The medulla is responsible for the secretion of epinephrine and norepinephrine, and would not be covered in this text. However, histological work showed that such strict anatomical distinction is not correct, as chromaffin cells were seen dispersed all over the cortex (Bornstein et al 1991 (1)
Adrenarche usually starts by the age of 8 years with increased production of DHEAS, followed after one year by increased production of androstenedione. There has been contradictory evidence regarding the initiating stimulus for adrenarche. It has been suggested that it could be affected through increase in body weight, and the related increase in insulin and leptin blood levels. Only a small role for ACTH has been confirmed in this respect (Apter et al 1979 (2), as there was no corresponding ACTH dependent increase in cortisol level as for the androgens. On the other hand, Weber et al (1997 (3) showed diminished adrenal androgens secretion in familial glucocorticoids deficiency, which they took as an indication for the importance of ACTH in the induction of adrenarche.
The zona glomerulosa is responsible for the production of aldosterone, where as cortisol is produced by the middle zona fasiculata. Adrenal androgens are produced mainly by the inner zona reticularis and partly by the fasiculata. Understanding the effects of the adrenal gland in reproductive medicine hinges on the knowledge of the different steps and enzymes involved in the production of the adrenal steroids. The two driving forces for the continued functioning of the adrenal cortex are aldosterone which is a mineralocorticoid, and cortisol which is a glucocorticoid hormone. Changes in the level of these two hormones are fine tuned in response to the body needs. Cortisol production is controlled by corticotrophin releasing hormone (CRH) and arginine-vasopressin (AVP), which is also known as antidiuretic hormone. They are both produced by the paraventricular hypothalamic nuclei. CRH is produced by the small or parvocellular neurosecretory cell into the portal circulation before reaching the adenohypophysis. This pattern is different to the AVP produced by the large or magnocelluar neurosecretory cells whose axons terminate in the infundibular process before being released into the general circulation. Aldosterone production is controlled by changes in the renin angiotensin system, with a minor role being played by ACTH. On the other hand, androgens are produced as byproducts during the production of these hormones, and their absence would not initiate any pituitary response to increase their production. At the same time they do not possess an active negative feedback mechanism to shut down their excessive production by the adrenal gland. This is because the rate limiting step in adrenal steroidogensis is triggered and slowed down according to the level of free cortisol in circulation.
Biochemistry
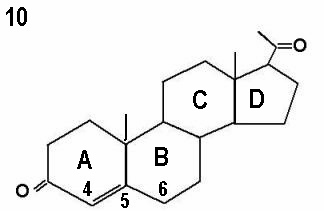
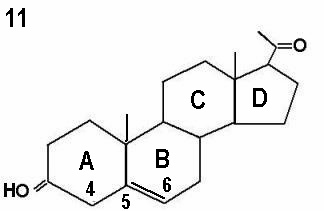
The initial step in the adrenal gland steroidal chains involves the conversion of cholesterol into pregnenolone by cholesterol side chain cleavage enzyme, catalysed by cytochrome P450scc. This is the rate limiting step, and is controlled by ACTH production by the pituitary gland. Almost 80% of cholesterol is retrieved as LDL cholesterol from the circulation, with only 20% synthesised locally. The next step involves further incorporation of pregnenolone into the D5 or D4 pathways. The term delta is used to indicate the location of the double bond within the steroid ring. The delta 4 molecules have a double bond between carbons 4 and 5 in ring A, whereas the double bond is between carbons 5 and 6 in ring B in delta 5 molecules. This is illustrated by the progesterone and pregnenolone molecules shown by Figures 10 and 11 respectively. Conversion of delta 5 precursors to delta 4 molecules is affected by the enzyme 3ß-hydroxysteroid dehydrogenase / delta 5 - delta 4 isomerase, which is also known as 3ßol-dehydrogenase (3ß HSD). Unlike other adrenal enzymes, it is not a member of the cytochrome p450 family.
 |
 |
The Delta 5 pathway
The final products of this pathway are weak androgens which could be converted to more potent one peripherally. Intermediate products could be converted into the D4 pathway as well. This conversion could be demonstrated as follow:
Pregnenolone » 17a-hydroxypregnenolone by 17a hydroxylase enzyme
17a-hydroxypregnenolone » dehydroepiandrosterone by 17/20 lyase enzyme
Dehydroepiandrosterone » androstenediol by 17b-hydroxysteroid dehydrogenase
The gene responsible for regulation of the 17a-hydroxylase enzyme is located in the long arm of chromosome number 10 (10q24) and is coded as CYP17A1.
The Delta 4 pathway
The initial step in this pathway is the conversion of pregnenolone and 17a-hydroxypregnenolone (which are D5 precursors) into progesterone and 17a-hydroxyprogesterone respectively by the enzyme 3b-hydrtoxysteroid dehydrogenase/D5-D4 isomerase. Defective conversion of the D5 precursors to D4 products could result in different grades of cortisol and aldosterone deficiency. In severe cases the newborn could have the classical or salt losing version with ambiguous genitals due to conversion of the accumulated D5 precursors peripherally to more potent androgens. The gene responsible for regulation of 3b-hydroxysteroid dehydrogenase enzyme is located in short arm of chromosome number 1 (1p13), and is coded as HSD3B2.
Cortisol production
The following steps are involved in cortisol production:
· 17a-hydroxyprogesterone » 11-deoxycotisol by 21 hydroxylase enzyme.
The gene controlling 21a-hydroxylase enzyme is located in the short arm of chromosome number 6 (6p21), and is given the gene symbol CYP21A2. Mutations in the function of this enzyme make more than 90% of all enzymatic deficiencies in the adrenal gland. The final outcome would be accumulation of the precursor 17a-hydroxyprogesterone ready for conversion into androgens as shown below, in the section dealing with androgens and oestrogens production. Severe deficiencies in this enzyme could lead to very low cortisol and aldosterone production with salt wasting and ambiguous genitals at birth. Milder forms could present after birth with symptoms mainly related to elevated androgens level. The next step in cortisol synthesis would be:
· 11-deoxycortisol » cortisol by 11b-hydroxylase enzyme. Mutations in the gene controlling this enzyme are the second most common after the 21a-hydroxylase type. They make about 8% of all adrenal enzymatic deficiencies.
The effective enzyme in this step is controlled by a gene located in the long arm of chromosome number 8 (8q21) and coded as CYP11B1. Gene mutation and partial block in this stage would lead to accumulation of 11-deoxycortisol and deoxycorticosterone. Severe enzymatic deficiency could also lead to ambiguous genitals at birth. Milder forms could cause hyperandrogenic symptoms and signs, as well as high blood pressure later on in life. The association of hypertension with low plasma renin activity follows deoxycorticosterone induced sodium retention and volume expansion. Hypokalaemia might also be an issue.
Aldosterone production
The following steps are responsible for the final production of aldosterone:
· Progesterone » deoxycorticosterone by 21a- hydroxylase enzyme
· Deoxycorticosterone » corticosterone by 11b-hydroxylase enzyme
· Corticosterone » aldosterone by aldosterone synthase enzyme.
It could be seen that the initial step in aldosterone synthesis shown above involves the enzyme 21a-hydroxylase enzyme which is also involved in cortisol synthesis. Deficiency in the production of this enzyme by the zona glomerulosa is not usually severe enough to cause major block in aldosterone production. However, with homozygotic patients such severe deficiency could occur, and lead to the classical salt wasting adrenal hyperplasia.
Androgens and oestrogens production
Androgens are byproducts and act as precursors to oestrogens as shown by the following steps:
· 17a-hydroxyprogesterone » androstenedione by 17/20 lyase enzyme
· Androstenedione » testosterone by 17b-hydroxysteroid dehydrogenase
· Androstenedione » oestrone by the aromatase enzyme
· Testosterone » oestradiol by the aromatase enzyme
The terms D5 and D4 denote the location of the double bond within the steroid nucleus. It has already been shown before that D5 precursors are converted to D4 products by the enzyme 3b-hydroxysteroid dehydrogenase.
Enzymatic Deficiencies
It is now easy to see how enzymatic blocks or deficiencies could halt the progress of the steroidal chain, and which precursor could build up behind that block. Theoretically, any of the enzymes involved in the steroidal chains could be deficient and lead to clinical effects depending on which hormone is deficient, and the alternative channels taken by the precursors at the bottleneck. Some of these deficiencies are not compatible with life, if not treated immediately, especially the rate limiting step involved with the conversion of cholesterol to pregnenolone. It is important also to say that different enzymes are controlled by genes located at different chromosomes. However, all mutation abnormalities involving the adrenal enzymes have autosomal recessive inheritance.
Total block of any enzyme would cause maximal effect. The time, mode and severity of manifestations depend on which hormone was deficient and the accumulated byproducts. Accordingly, enzymatic deficiencies could be categorised into four subgroups according to their severity:
- The classical or salt wasting types follow severe enzymatic deficiency leading to aldosterone and cortisol deficiency and excessive androgen production. This could follow severe deficiencies of 21a-hydroxylase, 11-hydroxylase and 3b-hydroxysteroid dehydrogenase. The affected neonate would also show different degrees of ambiguous genitalia. The HPA axis starts functioning by the 6th week of intrauterine life and malfunctioning of the adrenals with excessive production of androgens would affect the urogenital sinus normal sexual differentiation which occurs few weeks later.
Excessive intrauterine exposure to androgens could also have many effects on the attitude of the exposed female during childhood and adulthood periods of life. This would be addressed in more detail later on.
2. The nonclassical salt sparing or simple virilizing type follows partial enzymatic blocks, not including aldosterone and could present at birth or any time thereafter. With 21a-hydroxylase deficiency, Orta-Flores et al (1976 (4) showed one system with 2 different active sites, one on progesterone only and a second one on either progesterone or 17a-hydroxyprogesterone indiscriminately. They suggested that both sites are defective in the salt-losing variety and only the second site in the non salt losing type.
3. Late onset or attenuated adult onset form could lead to only hirsutism with no other signs in the most attenuated form.
4. Cryptic type in patients with biochemical evidence of 21a-hydroxylase deficiency without clinical signs of hyperandrogenisation, amenorrhoea or infertility (Levine et al, 1980 (5).
Specific types of enzymatic deficiency
21 alpha -hydroxylase deficiency
This is the most common type and makes >90% of all cases, as mentioned previously. Gynaecologists are more likely to deal with this version than the other types, and would accordingly be described in more detail. The main biochemical diagnostic parameter is a high blood level of 17a-hydroxyprogesterone. This could be tested in a morning blood sample in the basal state, or combined with an ACTH stimulation test. In the later scenario, 17a-hydroxyprogesterone level should be measured before and 60 minutes after a bolus dose of ACTH. An exaggerated increase in the level of 17a-hydroxyprogesterone would confirm the diagnosis. Like all other enzymatic deficiencies, the time and severity of presentations depend on the degree of the enzymatic block.
Neonatal manifestations
Normal development of the external genitals depends on the presence or absence of a testicle and the production of testosterone and dihydrotestosterone. More information has already been given in chapters 2 and 3. Enzymatic deficiencies however could affect the adrenal glands and gonads at the same time. Accordingly, when dealing with a newborn with ambiguous genitals the provisional diagnosis should include the following:
- Undermasculinized male
- Masculinized female
- True hermaphrodite
This is an emergency situation in the labour room which would cause great anxiety and distress to the parents. The condition is usually handled by a paediatric endocrinologist and later on by a paediatric surgeon if necessary. In these cases, exclusion of the classical type of 21a-hydroxylase deficiency with aldosterone deficiency should take priority, because of the difficult electrolytes deficiencies which could be fatal. However, the level of 17a-hydroxyprogesterone might give false results if tested within the first 24 hours after birth. It has been mentioned before that severe deficiency of 11b-hydroxylase and 3b-hydroxysteroid dehydrogenase could give a similar picture at birth. The masculinized vulva would take one of the following 5 stages as described by Prader as far back as 1955 (6):
- Cliteromegaly with no labial fusion
- Cliteromegaly with posterior labial fusion
- Larger clitoris with single perineal urogenital orifice with almost complete fusion of the labia
- Phallic clitoris with urethra-like urogenital sinus at its base and complete labial fusion
- Penile clitoris with urethral meatus at its tip and scrotum like labia.
Confirmation of the biochemical abnormalities and peripheral karyotyping is necessary to exclude the possibility of an undermasculinized male. The later anomalies could follow partial deficiency of the hormone 5a-reductase necessary for the complete masculinization of the urogenital sinus. Partial receptor insensitivity could also present in a similar manner. Patients with mixed gonadal dysgenesis should also be considered in the provisional diagnosis.
Childhood and Pubertal period
Females with the classical types of adrenal enzymatic deficiency would be outside the remit of gynaecologists, unless surgery is involved when gynaecologists experienced in this field would be involved. During childhood years, diagnosed cases of the less severe form of the enzymatic deficiencies could have slow growth and development. They may then suddenly show accelerated growth when the adrenals start producing excessive amounts of androgens at premature adrenarche. Children who are overdosed could show signs of underdevelopment as well and might even show some cushingoid feature due to excessive glucocorticoid replacement.
Within the gynaecological remit, children with the milder form might have premature adrenarche and start heterosexual precocious puberty before the age of 8 years. Parents might notice change in their daughters body odour due to early activation of the apocrine axillary sweat glands. There is also premature development of pubic hair and rapid linear growth. The child would be taller and more mature than her siblings. She might also show signs of skin hyperandrogenisation mainly acne with excessive facial or body hair. However, the final height would be shorter than expected. This is the usual final outcome despite meticulous control, in many cases. With excessive androgen production menarche might be delayed and the patient might even have primary amenorrhoea. Treatment with hydrocortisone or any other glucocorticoid should be started early and titrated against the level of the precursor hormone, mainly 17a-hydroxyprogesterone and androstenedione. Patients with 21a-hydroxylase deficiency are likely to develop polycystic ovaries secondary to the increased adrenal androgens production. This could lead to anovulation and abnormal uterine bleeding. Finally, infertility could be an issue and induction of ovulation might be needed despite good adrenal control with hydrocortisone which is preferable to dexamethasone or prednisolone in patients who would like to conceive as it is inactivated in the placenta and the fetus is not exposed to any exogenous steroids.
11beta-hydroxylase deficiency
Deficiency of the less common 11b-hydroxylase enzyme could follow a similar pattern of severity and association to aldosterone and 21a-hydroxylase deficiency. This could be in a classical or non-classical form as mentioned before. It could lead to different grades of hyperandrogenisation either at birth or at any other time, with high blood pressure on occasions but not in all cases. Diagnosis is usually confirmed by having a high 11-deoxycortisol blood level. Occasionally, a definitive diagnosis might be difficult and a stimulation test with metyrapone would be necessary. This would cause further block of the enzyme 11b-hydroxylase and stimulates ACTH production. An exaggerated increase in the level of 11-deoxycortisol would clinch the diagnosis. Patients with this enzymatic deficiency might have different health related problems due to the high blood pressure which could be severe at times. Treatment also involves hydrocortisone replacement therapy titrated against the level of 11-deoxycortisol.
17alpha-Hydroxylase Deficiency
Patients presenting with 17a-hydroxylase deficiency would have delayed or no signs of pubertal development, and primary amenorrhoea. This is secondary to failure of conversion of progesterone to 17a-hydroxyprogesterone and pregnenolone to 17a-hydroxypregnenolone. These are the precursors to androgens and later on oestrogen as shown before. Deficiency of the commonly associated 17/20 lyase enzymatic activity would compound matters further by reducing the conversion of the available 17a-hydroxyprogesterone and 17a-hydroxypregnenolone to androgens which are the substrates for oestrogens. They could also show high blood pressure and hypokalaemic alkalosis due to the conversion of the excessive progesterone to deoxycorticosterone and corticosterone. The age of onset and degree of severity of hypertension could vary between different individuals (Rosa et al 2007 (7).
A different biochemical picture would be seen in this type of enzymatic deficiency compared to the first 2 versions. High blood levels of FSH, LH, progesterone, deoxycorticosterone and corticosterone but low oestradiol level would be seen. Aldosterone and plasma renin activity would also be reduced. The ovaries might show multicystic pattern but ovarian biopsies showed no evidence of follicular maturation. Treatment would entail oestrogen replacement therapy to allow normal development of secondary sexual characteristic.
Other considerations
It is more common for gynaecologists to deal with teenage girls or young adult women who present with irregular periods and hyperandrogenic symptoms or signs. Even with the ultrasound diagnosis of polycystic ovaries, at least the level of 17a-hydroxyprogesterone should be tested to check for adult onset 21a-hydroxylase deficiency. Other than the ambiguous genitalia and precocious puberty alluded to before, high androgen levels can lead to primary or secondary amenorrhoea. Skin hyperandrogenization including acne, excessive hair growth of facial and body hair in a male pattern distribution, androgenic alopecia and hair recession may be a real problems. More severe hyperandrogenic or virilizing signs are seen only in severe cases. Ovulation dysfunction including, inadequate ovulation, short luteal phase with polymenorrhoea, menorrhagia, oligomenorrhoea and dysfunctional uterine bleeding are also common. Infertility can be an important issue to deal with due to the following detrimental effects of the high androgens:
-
- Polycystic ovarian conversion
- Reduced oocytes maturation capacity
- Reduce granulosa cells mitotic activity
- Reduced FSH induced aromatase activity
- Reduced follicular response to ovulation induction with clomid or gonadotrophins
- Affect the endometrium and reduce pregnancy rate
Intrauterine exposure to high levels of androgens has been shown to affect the gender behaviour and attitude of exposed females both as children and during their adult life. They could be less tender minded, with greater physical aggression tendency and show less interest in infants (Mathew et al (8). They were also shown to have less heterosexual interest and were not satisfied with their sexual assignment as females. However, these changes were different across different behaviours in comparison to normal controls (Sheri et al 2003 (9). Accordingly, it is not possible to predict across different behaviours in this complex subject. Nevertheless, it is generally agreed that severe intrauterine exposure to androgens, especially with the classical types of adrenal hyperplasia, is more likely to cause behavioural masculinization during childhood and adult life.
Normal menstruation usually resumes some time after normalisation of 17a-hydroxyprogesterone in patients affected by 21a-hydroxylase deficiency. Treatment with dexamethasone 0.25-0.5 mg every night for 1-2 years could results in normal ovulatory cycles even after stopping medication (McKenna 1991 (10). However, some patients might remain to have persistently high serum progesterone level despite the correction of the 17a-hydroxylase level (Helleday and Holmes-Walker 1993 (11). This could lead anovulatory cycles (Nimkarn and New 2008 12), or failure of the endometrium to develop despite good follicular maturation (Holmes-Walker et al 1995 (13).
Family considerations
All adrenal enzymatic deficiencies are inherited as autosomal recessive disorders. With carrier parents, one child would be affected (25% chance), two would be carriers (50% chance) and one would miss the abnormal genes (25% chance). Severity of the mutation would also affect the penetration. A severely affected allele could be inherited as a severe abnormality. Premarital or preconception screening might be useful, especially if one prospective parent proved to be a carrier. This is especially so in communities known for intermarriages or marriages within the same ethnic group. Similarly, a female sibling of a hyperandrogenic patient diagnosed with such adrenal defect should be offered proper screening, especially if she showed the same symptoms or signs.
Parents of a previously affected child or carrier parents might seek preconception advice. Two methods could be used to deal with this problem including Preimplantation genetic diagnosis and prenatal diagnosis.
1. Preimplantation genetic diagnosis (PGD)
With PGD, in vitro fertilisation would be necessary to create embryos which should be biopsied and tested for the enzymatic deficiency. Only normal embryos should be replaced into the patients uterus. This might create ethical issues of creating embryos and then destroying them. Normal embryos could be destroyed during the procedure which is not 100% accurate. On the other hand, such screening would allow parents a better chance of having healthy children and could prevent the potential sufferings of the affected children. There is also the option of prenatal diagnosis which could be too late to make a diagnosis in time to prevent virilization of a female fetus. Accordingly, thorough information counselling would be necessary and patients would need time to make their own minds without any pressure. The availability of the service does not indicate forcing parents to use it.
2. Prenatal genetic diagnosis (CVS)
Prenatal molecular genetic studies of extracted fetal DNA retrieved by chorionic vellus sampling are the ideal methods for diagnosing 21a-hydroxylase deficiency (White and Speiser, 2000 (14). Affected pregnancies should be supplemented with dexamethasone. However, this could be rather late to prevent exposure of the urogenital sinus to the excessive fetal androgens, bearing in mind that the fetal adrenal gland starts functioning by the 6th week of pregnancy, well before a CVS could be done. Accordingly, dexamethasone should be started as soon as pregnancy is diagnosed and not later than by the 9th week as advised before Mercado et al (1995 (15) & Carlson et al (1999 (16). CVS could be done as usual, and medication could be stopped if the fetus proved to be normal. The recommended dose of dexamethasone is 20 µg /kg pre-pregnancy weight / day in divided doses.
Summary
This short manuscript was written to address the problems posed by dysfunctional adrenal glands as seen by gynaecologists. The relationship between adrenal dysfunction and the risks of ambiguous genitalia, abnormal pubertal development, anovulation, dysfunctional uterine bleeding, hyperandrogenic skin manifestations and infertility are too many to be ignored by any gynaecologist. Accordingly, assessment of 17a-hydroxyprogesterone should be included during the investigations of all these conditions to exclude this often forgotten problem.
Reference
1. Bornstein SR, Ehrhart-Bornstein M, Usadel H, Bockmann M, Scherbaum WA. Morphological evidence for a close interaction of chromaffin cells with cortisol cell within the adrenal gland. Cell Tissue Res 1991; 265: 1-9.
2. Apter D, Pakkerinen A, Hammond GI, Vihko R: Adrenocortical function and puberty, serum ACTH cortisol and dehydroepiandrosterone in girls and boys. Acta Pediatr Scand 1979; 69: 599 - 606.
3. Weber A, Clark AJ, Perry LA, Honour JW, Savage MO: Diminished adrenal androgen secretion in familial glucocorticoid deficiency implicates a significant role for ACTH in the induction of adrenarche. Clin Endocrinol (Oxf) 1997; 46: 431- 437.
4. Orta-Flores Z, Cantu JM and Dominguez MB. Reciprocal interactions of progesterone and 17 alpha-OH-progesterone as exogenous substrate for rat adrenal 21-hydroxylase. J Steroid Biochem 1976; 7: 761 - 767
5. Levine LS, Dupont B, Lorenzen F, Pang S, Pollack M, Oberfield S, Kohn B, Lerner A, Cacciari E, Mantero F, Cassio A, Scaroni C, Chiumello G, Rondanini GF, Gargantini L, Giovannelli G, Virdis R, Bartolotta E, Migliori C, Pintor C, Tato L, Barboni F and New MI Cryptic 21-hydroxylase deficiency in families of patients with classical congenital adrenal hyperplasia. J Clin Endocr Metab 1980; 51: 1316 - 1324.
6. Prader A and Gurtner HP. The syndrome of male pseudohermaphrodism in congenital adrenocortical hyperplasia without overproduction of androgens (adrenal male pseudohermaphrodism. Helv Paediatr Acta 1955; 10(4): 397 - 412.
7. Rosa S, Duff C, Meyer M, Lang-Muritano M, Balercia G, Boscaro M, Kemal Topaloglu A, Mioni R, Fallo F, Zuliani L, Mantero F, Schoenle EJ and Biason-Lauber A. P450c17 Deficiency: Clinical and Molecular Characterization of Six Patients. The Journal of Clinical Endocrinology & Metabolism 2007; 92(3): 1000 - 1007
8. Mathews GA, Fane BA, Conway GS, Brook CGD and Hines M. Personality and congenital adrenal hyperplasia: Possible effects of prenatal androgen exposure Hormones and Behaviour 2009; 55(2): 285 - 291.
9. Sheri A. Berenbaum and J. Michael Bailey Effects on Gender Identity of Prenatal Androgens and Genital Appearance: Evidence from Girls with Congenital Adrenal Hyperplasia. J Clin Endocrinol Metab 2003; 88(3): 1102 - 1106.
10. McKenna TJ. The treatment of chronic hyperandrogenemic states: adrenal suppression. In Coelingh Bennik HJT, Vermer HM and van Keep PA, eds. Chronic hyperandrogenic anovulation, Carnforth, UK: Parthenon Publishing, 1991: 143 148.
11.Helleday J, Siwers B, Ritzén EM and Carlström K. Subnormal androgen and elevated progesterone levels in women treated for congenital virilizing 21-hydroxylase deficiency. J Clin Endocrinol Metab 1993; 76 (4): 933 - 936.
12.Holmes-Walker DJ, Conway GS, Honour JW, Rumsby G and Jacobs HS. Menstrual disturbances and hypersecretion of progesterone in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol 1995; 43 (3): 291 - 296.
13.Nimkarn S and New MI. Steroid 11 beta-hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab 2008; 19 (3): 96-9.
14.White P and Speiser P. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev 2000; 21: 245 - 291.
15.Mercado AB, Wilson RC, Cheng KC Wei JQ and New MI. Extensive personal experience: Prenatal treatment and diagnosis of congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. J Clin Endocrinol Metab 1995; 80: 2014 - 2020.
16.Carlson AD, Obeid JS, Kanellopoulou N, Wilson RC and New MI. Congenital adrenal hyperplasia: update on prenatal diagnosis and treatment. 10TH International Congress on Hormonal Steroids, Quebec, CA, 1999; pp 19 - 20.
|




