| |
Chapter 5 Progestogens, Androgens and Oestrogens
This chapter has been written to give a functional account of
progestogens, androgens and oestrogens, rather than a biochemical
script to compare their structural similarities and differences.
Understanding their metabolic and morphological effects and their
interactions with each other and with other endocrine glands forms the
final objectives behind writing this chapter.
Progestogens, androgens and oestrogens are steroidal compounds made
of 4 interconnected cyclic hydrocarbons rings designated as A, B, C and
D rings respectively. They can be natural or synthetic. All steroidal
hormones attach to intracytoplasmic receptors, before being carried into
the nuclei to exert their specific effects. Different hormones have different
potency, depending on the duration of time a single dose of steroid-
receptor complex occupies the nucleus of the target cell. In their natural
forms, they are produced by the adrenal glands and ovaries in non
pregnant women. Peripheral conversions also play an important role in
the synthesis and degradation of oestrogens and androgens. The
placenta is a major source during pregnancy
Progestogens
Progesterone was discovered in 1934. It is a natural 21-carbon
molecule formed from pregnenolone by the microsomal enzyme 3β-
hydroxysteroid dehydrogenase/Δ5-Δ4 isomerase. Pregnenolone itself is
made from cholesterol through a reaction catalysed by cytochrome
P450scc, as described in Chapter 4. In a way, progesterone is a mother
molecule of androgens and oestrogens, as shown by the following
chart:
Progesterone → 17α-hydroxyprogesterone ↓
Androstenedione → Oestrone ↓
Testosterone → Oestradiol
The importance of this interrelationship has been discussed in
Chapter 4, where failure of conversion of progesterone to 17α-
hydroxyprogesterone can lead to failure of androgens and oestrogens
production.
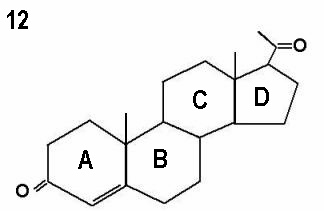
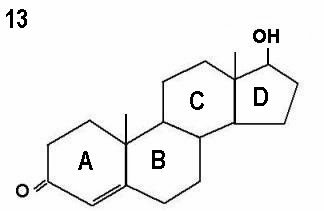
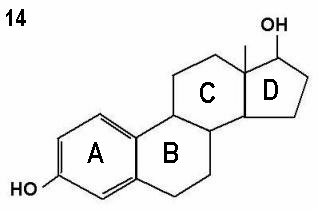
The three molecules shown in Figures 12 - 14 represent progesterone,
testosterone and oestradiol respectively. Note the small differences in the basic
two dimensional structures of the three molecules.
Natural progesterone is produced by the corpus luteum, adrenal glands
and the placenta. Synthetic progestogens, on the other hand, are
mainly the derivatives of:
- 17α-hydroxyprogesterone which are non-androgenic;
- 19-norprogesterone derivatives which are also non-androgenic;
- 19-nortestosterone derivatives which are androgenic.
Natural progesterone is >95% bound to plasma proteins, mainly
albumen and transcortin. Once in the blood, it has a short half-life of 5-
20 minutes. Accordingly, the efficacy of exogenous progesterone
depends more on the route of administration and its absorption half-
life, rather than its elimination rate. It is metabolised in the liver
successively into pregnanedione, pregnolone, and finally pregnandiol.
The effects of natural progesterone can be divided into the following
categories:
1. Endocrine or chemical effects Following ovulation, progesterone produced by the corpus luteum
modulates the function of the hypothalamo-pituitary units. It affects
GnRH pulse generation leading to slower LH pulse pattern with high
amplitude during the luteal phase. The central effect of
progesterone is affected at the level of the hypothalamus itself (1).
Patients with reduced hypothalamic sensitivity to progesterone will
have rapid GnRH and LH pulses, as seen in patients with polycystic
ovary syndrome (2). It depletes oestrogen receptors as well as its own. This is
important at the endometrial level, as prolonged endometrium exposure to progesterone can lead to endometrial atrophy, and
possibly dysfunctional uterine bleeding. It also affects oestradiol
metabolism by increasing its conversion to oestrone. This is
affected through activation of the enzyme 17-hydroxysteroid
dehydrogenase. It competes weakly with testosterone at its receptors level.It has a thermogenic effect by increasing the core body temperature during the luteal phase. 2. Morphological effects
-
The immediate morphological effect of progesterone after
ovulation is to increase cervical mucous viscosity, which reduces
migration of bacteria and sperm into the cervical canal.
-
It converts the oestrogen primed endometrium into a secretory
one. This is one of the most commonly used indications for
progesterone medication. It can be given by deep intramuscular
injections, or vaginally during fertility treatment, especially with
assisted reproduction. Cyclogest pessaries and crinone gel are just
two examples in common use. A meta-analysis published by
Zarutskiea and Phillips in 2007 (3) showed that transvaginal
progesterone medication in the right daily dosage is equally
effective as the intramuscular route in this respect. Micronization
of progesterone increased its surface area, and improved its
absorption through the stomach. It has been licensed by the
American Food and Drug Administration (FDA) for the
management of secondary amenorrhoea and for hormone
replacement therapy since 1998.
-
It has an effect on tubal peristaltic activity, and reduces the
number of cilia and mucous production by the fallopian tubes.
-
The antioestrogenic effect of progesterone has a direct effect in
reducing myometrial sensitivity and contractility.
It augments the effect of prolactin in preparing the breasts for
lactation, but also inhibits lactation during pregnancy. The
dramatic decline in the blood level of progesterone following
childbirth triggers milk production.
3. Metabolic and immunological effects
Progesterone has an immunosuppressant effect which is not
mediated through progesterone or glucocorticoid receptors. This
effect was thought to be secondary to non-receptor mediated
activity, conversion of progesterone to another steroid within
the microenvironment of the immune cell, and interaction of progesterone with other members of the steroid and thyroid hormone receptor superfamily (4). - It has a catabolic effect, as shown by a rapid decline in the plasma concentration of many amino acids after progesterone
administration (5). There is also increased total urinary nitrogen
excretion without aminoaciduria.
- It may induce hyperinsulinaemia by acting directly on the
pancreas and promotes hepatic storage of glycogen.
- Its also
antagonises the effect of insulin on glucose metabolism in adipose
tissues and muscles (6). This is coupled by increased deposition
of fat in the breasts and adipose tissue.
- It also reduces the
hypertriglyceridaemic effect of oestrogen.
- It has an anti aldosterone effect, and increases sodium loss.
- It increases the respiratory minute tidal volume, hence reduces the alveolar and blood CO2.
Synthetic progestogens Synthetic progesterone analogues have been produced, as
progesterone is poorly absorbed from the gastrointestinal tract unless
it is micronised Such progestogens have different structures and
characteristics, but they all share the common ability to induce
secretory changes in an oestrogen primed endometrium. Unlike natural
progesterone, many of them can be taken orally. Like natural
progesterone, they modify oestrogen effects but have different
characteristics otherwise:Progestogens derived from 19-nortestosterone have different
degrees of androgenicity. The sequence of ascending androgenicity
is: ethynodiol diacetate, norethindrone, norethindrone acetate,
norgestimate and desogestrel in thatorder, with levonorgestrel
and gestodene having the highest androgenicity. Derivatives of 19-norprogesterone are referred to as pure
progestational molecules, as they have no androgenic,
oestrogenic or glucocorticoid activity. They bind almost
exclusively to progesterone receptors. This group includes
nestorone, trimegestone and nomegestrol (7).Mild corticoid effect has been attributed to cyproterone acetate
(8), but it also competes with cortisol at its receptor sites (9).
Furthermore, it has mild inhibitory effect on the enzyme 21-
hydroxylase and to a lesser extent 3βol-dehydrogenase (10).
Accordingly, it can inhibit the production of both cortisol and
aldosterone at the same time. The degree of inhibition is dependent on the metabolic clearance rate of the drug, and the
degree of the genetic mutations of the two enzymes in the
individual concerned. Because of adrenal glands suppression and
their reduced response to ACTH, cyproterone acetate should be
withdrawn slowly to prevent adrenal failure, especially if it has
been used in a high dose for a long period of time. The new progestogen drospirenone is a derivative of
spironolactone, and has an anti mineralocorticoid effect.
Accordingly, it decreases salt and water retention with a potential
for lowering blood pressure (6). It also has a mild antiandrogenic
effect. Progestogens have anti gonadotrophin effect when given in a high
dose. Most progestogens suppress the production of endogenous
progesterone by affecting the corpus luteum enzymatic activity, if
used during the luteal phase. There is no actual luteolytic
activity, as suppression can be reversed by human chorionic
gonadotrophin injections. The lowest total dose necessary to
produce such an effect was 30 mg for northisterone, 12 mg for
norgestrel, 300 mg for chlormadinone acetate and 360 mg for
medroxyprogesterone acetate (11).
Most synthetic progestogens are derived from testosterone, especially
those used in oral contraceptives. More information about their
biochemical subgrouping will be found in Chapter 13. They have different
effects on lipids and lipoproteins, depending on their androgenicity and
the dosage used (12). They increase LDL production, but increase its
clearance rate as well. Accordingly, they have no significant effect in this
respect when used with oestrogen. Androgenic progestogens, such as
levonorgestrel, decrease triglyceride levels by reducing secretion of very-
low density lipoprotein (VLDL). On the downside, they can also
counteract the beneficial effect of oestrogen on HDL (13). Conversely,
non-androgenic progestogens have variable effects on oestrogen induced
increase in HDL level. Dydrogesterone, as an example, has little negative
effect (14), whereas medroxyprogesterone acetate reduces this effect. In
general, C-21 progestogens do not prevent the increase in triglycerides
induced by oral oestrogens.
The derivatives of 17α-hydroxyprogesterone and 19-norprogesterone
are antioestrogenic and antigonadotrophic. They have no androgenic
effect, which makes them favourable to use in patients with
hyperandrogenic tendency. They can be given orally or by injection. The
two main subgroups of 17α hydroxyprogesterone are: - 17α-hydroxyprogesterone caproate which is given
intramuscularly;
- 6α-methyl-hydroxyprogesterone acetate which is also known as
medroxyprogesterone acetate or provera can be used orally. It
can also be used intramuscularly in a depo form which increases
its duration of action (depo-medroxyprogesterone acetate).Depo-medroxyprogesterone acetate is used mainly as a long acting
injectable contraceptive, and for ovulation suppression in cases of
endometriosis. The term medroxy is an abbreviation for methyl-
hydroxy. Deep intramuscular injections can be repeated every 3
months in doses of 150 mg. More frequent injections do not improve
the efficacy of the drug, but may result in more side effects. The main
side effects even when the drug is used in the recommended doses
include:Prolonged amenorrhoea may occur even after suspending
medication. The average period for the return of normal fertility
has been quoted as 9 months (15), but it may take even longer
time after prolonged use of the drug.
- Risk of abnormal uterine bleeding is also an issue. Prolonged
periods of bleeding both heavy and light may be encountered.
- There is a risk of developing ovarian cysts.
- The prolonged anti oestrogenic effect on the brain may lead to lower mood spells, and occasionally depression.
- Other anti oestrogenic side effects may be a problem, mainly osteoporosis.There is also a risk of weight gain after prolonged use of the drug.
Other long acting 17-hydroxyprogestogen derivatives have been
used for:
-
Supplementing pregnancy following repeated miscarriages and
premature labour has been one indication for using 17-
hydroxyprogesterone caproate. The brand mostly used was
Delalutin, which has been withdrawn in 1999. In 2008, the
American FDA regarded 17-hydroxyprogesterone caproate as a
category D drug, which indicated evidence of fetal harm, when this
drug was used during pregnancy. Nonetheless, many articles have
defended the safety of 17- hydroxyprogesterone during pregnancy
on theoretical basis, as it is produced in large amounts by the
placenta, and according to the results of animal and clinical studies.
Most of these publications dated back to the 60s, 70s and 80s.
-
A well known member of this subgroup is cyproterone acetate.
It has a very potent anti androgenic effect by competing with testosterone and dihydrotestosterone at their receptors. It also
has a strong antigonadotrophic effect. It is most commonly used
for treatment of female hyperandrogenisation in a reversed
sequential therapy in severe cases. Because of its depo effect, it
may lead to menstrual irregularities or dysfunctional uterine
bleeding, unless it is combined with an oestrogen. It can be used
in a dose of 10-50 mg every day plus 30 μg of ethinyl oestradiol
for 10 days, to be followed by unopposed ethinyl oestradiol for 15
days. In milder cases, it can be used in a small dose of 2 mg
combined with 35 μg ethinyl oestradiol in a designated
contraceptive pill called Dianette (Bayer plc). Cyproterone acetate
can cause breast tenderness, lethargy, depression, loss of libido
and adrenal suppression. An important side effect is dysfunctional
uterine bleeding. Accordingly, it should not be used in the second
half of the cycle. The objective of using oestrogen is to regulate
the monthly withdrawal bleeding. Cyproterone acetate is
contraindicated in cases of liver disease, severe depression,
history of deep vein thrombosis and during pregnancy.
In most cases, further progestogen medication will not stop progestogen
induced abnormal uterine bleeding, and may even make it worse. This is
especially so after using long acting progestogens. During mild to
moderate bleeding episodes, oral oestrogen helps in building up the
endometrium and gives it some structural integrity, before bleeding
stops. In severe cases, only intravenous oestrogen may be effective.
Premarin can be used in a dose of 25 mg every 4 hours for 24 hours,
followed by oral oestrogen for further 10 14 days. A progestogen is
needed during the last 5-7 days of therapy to induce secretory
endometrial changes, before suspending treatment to provoke a
medically controlled withdrawal bleeding. Blood loss usually eases off
substantially, or even stops after the 3rd or 4th premarin intravenous
dose. Progestogens are also useful for treating patients with anovulatory
irregular or abnormal uterine bleeding. However, luteal progestogens
medication is not useful for controlling ovulatory abnormal uterine
periods. They may even be detrimental, and cause more menstrual blood
loss (16; 17). In contrast, longer regimens from days 5-25 of the cycle
are equally effective as levonorgestrel intrauterine devices, but only have
30% acceptability by patients for repeated prescriptions (18).
Transvaginal ultrasound scan examination can be very useful in setting
the management plan. Patients presenting with abnormal uterine
bleeding and a thin endometrium should have oestrogen to build up the
endometrium, and to benefit from its local haemostatic effect. The
indiscriminate use of progestogens for all patients should be avoided.
Androgenic progestogens are mainly testosterone derivatives, after
removal of the C19 methyl group (19-nortestosterone). The most
commonly used one outside the contraception field is norethisterone
(primolut N) which is a 17α-ethinyl derivative of 19-nortestosterone. It
is mainly used for inducing withdrawal bleeding after periods of
amenorrhoea, and for treatment of anovulatory menorrhagia. The
other commonly used derivative is 13 ethinyl-17α-ethinyl-19
nortestosterone (norgestrel). This and other androgenic progestogens
are mainly used in minute amounts either as progestogen only pills, or
as part of combined contraceptive pills. The mirena system is a
levonorgestrel loaded intrauterine contraceptive device which is used
for contraception, control of excessive uterine bleeding, and in cases of
endometriosis for pain control. It is impregnated with 52 mg of
levonorgestrel, and releases 20 μg of the hormone into the uterine
cavity every day. Only a small fraction reaches the general circulation,
though it has been detectable in significant amounts in the peritoneal
fluid in the pelvis. It has also been shown to have anti-inflammatory
and immunomodulatory effects (19). Furthermore, levonorgestrel
decreases and then blocks DNA synthesis and mitotic activity (20). In
addition, it increases endometrial apoptotic activity by reducing
expression of the Bcl-2 gene which has an anti-apoptotic effect (21).
It is advisable to avoid androgenic progestogens use in hyperandrogenic
women, as they may worsen this condition. This is especially so for
norgestrel and levonorgestrel as they can suppress the production of sex
hormone binding globulin by the liver, and increases the level of free
testosterone. High doses of norethisterone acetate for long periods of time
in repeated cycles, to control abnormal uterine bleeding, are better
replaced with medroxyprogesterone acetate which is equally effective
when used in the right dose. Furthermore, many 19-nortestosterone
derivatives are capable of reducing HDL cholesterol level, and inducing
insulin resistance. This is also valid for gestational diabetes mellitus
(GDM), as shown by Hedderson et al in 2007 (22). Their results suggested
43% increased risk of GDM associated with pre-pregnancy use of high
androgen hormonal contraceptives.
Human testing of progestogens
Progestogens potency was measured in different ways including: - The progestational dose is the one capable of converting an
oestrogen primed endometrium into a secretory one. Secondary
amenorrhoeic women were prescribed unopposed oestrogen for two weeks, followed by combined oestrogen and progestogen
medication for 10 days. The endometrium by the end of this
period was assessed for different grades of secretory changes.
The limitations of this test were discussed by Swyer in 1984 (23),
who argued that no data satisfied the standards of acceptability,
on his opinion, by the time his paper was published.
- The cycle delaying dose was documented first by Greenblatt et al
in 1958 (24). It is the progestogen dose capable of delaying
menstruation when started 7 days after ovulation, and continued
for 3 or more weeks. Bleeding should only start 2-3 days after
stopping the effective progestogen therapy, and not during
medication.
- Other parameters used to test progestogens potency included
depression of the vaginal karyopyknotic index, inhibition of
oestrogen induced cervical mucous changes, inhibition of
ovulation, and withdrawal bleeding after 5-days courses in
women with secondary amenorrhoea and oestrogen primed
endometrium. These tests were utilised as a guide for selecting the right doses of
progestogens to be used in new contraceptives, with standardized
doses of ethinyl oestradiol. They are also helpful in selecting the
effective dosage to control abnormal uterine bleeding. They should not
be used for delaying menstruation for social or religious occasions.
Such practice may lead to abnormal uterine bleeding, especially if the
correct cycle delaying dose is not used.
Table 1: shows the total progestational dose (TPD), and
the daily cycle delaying dose (CDD) of tested progestogens
|
Progestogens
|
TPD
|
CDD
|
|
Pure progesterone im
|
200 mg
|
1000 mg
|
|
Northisterone
|
100-150 mg
|
15 mg
|
|
Medroxyprogesterone acetate
|
60 mg
|
20 mg
|
|
Cyproterone acetate
|
20 mg
|
Not tested
|
|
Levonorgestrel
|
7 mg
|
5 mg |
Androgens
Androgens are C19 steroidal hormones which are capable of initiating
and maintaining secondary male sexual characteristics. They may be
natural or synthetic in origin. They are also capable of inducing nitrogen
retention, and have high affinity to certain cytoplasmic prostate cell
receptors. They are produced in women by the ovaries, adrenal glands, and by peripheral conversion in the skin, fat, and by the liver. The two
main circulating androgens are testosterone and androstenedione (Δ4-
A). However, in a decreasing order of production in adult women, the
major androgens are dehydroepiandrostenedione sulphate (DHEAS),
dehydroepiandrostenedione (DHEA), androstenedione, androstandiol
(Δ5A-diol), testosterone and dihydrotestosterone (DHT) (25). At skin
level, dihydrotestosterone is the main functional androgen molecule,
and is made by peripheral conversion from androstenedione (70%) and
testosterone (20%), as well as other precursors by the enzyme 5α-
reductase. Such conversion is not required at other tissues including the
brain or muscles, as testosterone is the main active molecule in these
sites. Only a small amount of dihydrotestosterone is actively produced
by the ovaries.
Normally, the ovaries and adrenals produce 20-25% of circulating
testosterone each, with the remaining 50% produced by peripheral
conversion of androstenedione. On the other hand 35% of circulating
androstenedione is produced by the ovaries and 25% by the adrenals,
with the rest through peripheral conversion. Almost 80% of circulating
testosterone is bound to SHBG, 19% to albumen and 1% is free as an
active fraction. The blood level of androgens depends on the production
rate, available SHBG receptor sites and the metabolic clearance rate by
the liver, skin, peripheral fat and other tissues.
Many routes are available for androgens metabolism. Peripheral fat and
muscles are two main tissues involved with aromatisation of androgens
to oestradiol and oestrone. Testosterone is also metabolised to
androsterone and aetiocholanolone in peripheral tissues, before being
conjugated in the liver to glucuronide and sulphate by-products. These
are water soluble and excreted in urine. Such conjugation occurs mainly
at C-17 and C-3 sites in the androgen molecules. In general hepatic
extraction of androgens is inversely related to their SHBG binding.
About 40-60 % of testosterone and 30-40% DHT are extracted by the
liver (26; 27). Generally androgens have the following functions in the human
female: - Classical teaching attributed the initiation of puberty and growth
in linear height to adrenal androgens. This concept has been
challenged recently, and both phenomena were related instead to
increased levels of oestrogen and growth hormone, as discussed
in Chapter 2.
- They are responsible for the growth of ambisexual axillary and
pubic hair, which needs only small amount of androgens.
- They act as substrate for oestrogens production.They are involved in maintaining female fertility, mainly through
regulation of the hypothalamo-pituitary axis in a dose dependantmanner (28);
- They play a role in regulating follicles development and ovulation at the level of the ovaries. This is partly affected through down
regulation of FSH receptors in the small follicles to promote
monofollicular ovulation.
- They can help in increasing libido at the time of ovulation.The biological activity of androgens is controlled mainly by their free
fraction in circulation which is controlled by their production rate,
metabolic clearance rate, and interaction with other hormones. In
normal circumstance, this is mainly affected by the level of SHBG
which is produced by the liver. SHBG production is increased by
oestrogens, thyroxine and reduced by obesity, hypothyroidism and
high androgens production. It protects androgens against rapid
degradation, and accordingly controls their metabolic clearance rate.
It also has a role in controlling interconversions between androgens,
and the peripheral conversion of testosterone to oestradiol. In this
respect, measurement of total testosterone does not reflect the
free and effective fraction during investigations of female
hyperandrogenicity. Accordingly, the testosterone / SHBG ratio,
usually known as the free androgen index, is a better measure of
clinically relevant androgenicity than the total testosterone level.
- Certain androgens have higher affinity to SHBG sites than others, and
displace testosterone from its binding sites, leading to high levels of the
biologically active free testosterone. Norgestrel is one example with
higher tendency in smaller doses than northisterone. Small doses of
300 μg of northisterone may not affect SHBG level, but daily doses of 5
mg, which are usually used to control abnormal uterine bleeding, can
do so. On the other hand, cyproterone acetate does not affect SHBG.
Timing the blood test for androgens level assessment is important in
relation to the time of the day, and relative to the menstrual cycle.
Androgens are produced in circadian pattern, especially the adrenal
ones, and are higher in morning blood samples. Afternoon samples may
give erroneously low values. Furthermore, blood samples should be
timed to the early or mid follicular phase, as testosterone level can be
high in the middle of the cycle. An example of such a scenario is thathigh LH and testosterone blood levels on days 5-7 of the cycle can
represent PCOS. In contrast, even higher LH and testosterone levels at
the middle of the cycle will be a normal physiological finding at the time
of the LH surge.
Excessive androgens may affect a female patient during the different
stages of her life differently, as follows:Excessive exposure of a female fetus to androgens during
intrauterine life can lead to ambiguous genitalia at birth. Similar
exposure of the fetal brain may adversely affect gender identity
during childhood and adulthood life. It can also impinge on the
heterosexuality of the adult woman.
Excessive androgens exposure during childhood can lead to
precocious heterosexual puberty as discussed in Chapter 2. It
may also cause primary or secondary amenorrhea and skin
hyperandrogenic signs. During adult life excessive androgens exposure leads to general
hyperandrogenic symptoms and signs including weight gain, acne,
hirsutism, androgenic alopecia, and other masculinization signs.
Severe cases will show virilization signs including cliteromegaly,
frontal hair recession and coarse voice. Occasionally, excessive
sexual hair growth and acne may occur despite normal levels of
circulating androgens. In such cases increased production of DHT
can follow high tissues 5α-reductase activity. This is reflected by
increased production of 5α-androstandiol-3α, 17β-diol glucoronide
which is a byproduct of DHT (29).
Excessive androgens can also affect the HPO axis and uterus, and
cause luteal phase defects, polymenorrhoea, menorrhagia,
dysfunctional uterine bleeding, oligomenorrhoea and amenorrhoea.
Detrimental direct effects at the level of the oocytes and
endometrium have also been confirmed. Androgens can reduce
oocytes maturation capacity, reduce granulosa cells mitotic activity,
and FSH induced aromatase activity.
Management of hyperandrogenaemic states The most important steps in the management of hyperandrogenaemia
are:
Stop any medication which can lead to hyperandrogenism.
Exclude adrenal or ovarian tumours as a cause, especially in
women with adults onset conditions, rapid progressive signs, and very high blood levels of androgens.
-
Stop or reduce the production of the androgens, being ovarian or
adrenal in origin. Different drugs are available for this purpose.
Adrenal enzymatic deficiencies are treated with glucocorticoids and
ovarian sources are managed with oral contraceptive pills, or
gonadotrophin releasing hormone analogues
-
Increase the level of blood SHBG which reduces the level of free
testosterone. This can be affected through the oestrogen fraction
of an oral contraceptive pill. It has been shown that 30 μg ethinyl
oestradiol did not increase SHBG level beyond levels seen in
normal ovulating women. The effect was more dramatic with 50
μg doses.
-
Antiandrogens should be used to counteract the effects of the
androgens on peripheral tissues. The most widely used drugs are
spironolactone and cyproterone acetate. They compete with
androgens at their receptor sites.
-
Patients with polycystic ovary syndrome may benefit from
metformin which can reduce ovarian production of excessive
androgens.
-
Skin care and proper use of cosmetic aids are important parts of
the management plan, to support the antiandrogenic treatment.
Assess the psychological impact of the problem and the basis for
presentation. Counselling may help as well.
Clinical use of androgens in female reproductive medicine is limited by
their side effects, which can be disfiguring and not acceptable. Danazol,
which is an isoxazole derivative of 17α-ethinyl testosterone, used to be
popular for the treatment of endometriosis. It has also been used for
the treatment of mastalgia in a daily dose of 50-100 mg during the
luteal phase. Testosterone implants were also used to supplement
oestrogen HRT in women with low libido, but are not popular now.
Recently, the transdermal route has been tested as well. Intrinsa
patches (Procter and Gamble) provide 300 μg of testosterone every
day, and each patch can be used for 3-4 days. They are licensed for
women with hypoactive sexual disorder on concomitant oestrogen
therapy, after bilateral oophorectomy. Beside acne, excessive hair
growth and weight gain, androgens can cause migraine, insomnia,
breast pain, and dyslipidaemia with low HDL cholesterol and high LDL
cholesterol.
Oestrogens
Oestrogens are biologically defined as chemicals which promote sexual
heat or oestrous in ovariectomised premature rats. They are also defined as substances that promote vaginal cornification or uterotropic
effects in the oophorectomised rat or mouse. In a clinical context,
oestrogens are known as chemicals that stimulate and maintain growth
of female secondary sexual characteristics. As for progestogens, they
are also classified as naturally occurring, and synthetic. It has been
shown earlier in this chapter that oestradiol and oestrone are steroidal
compounds produced from androgens, both in the ovaries and adrenal
glands in non pregnant women. Peripheral conversion of androgens in
the skin and fat also contributes to the level of these two hormones. The
placenta is a major source of oestriol during pregnancy.
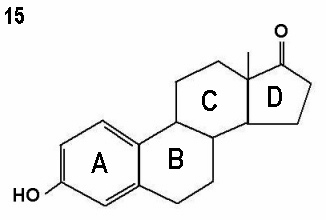
Structurally oestrone, oestradiol and oestriol have 18 carbon atoms
each, but differ in the number of the hydroxyl groups within the
molecule.
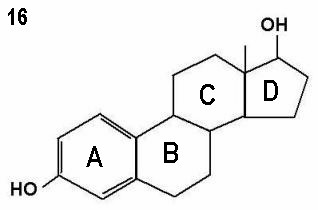
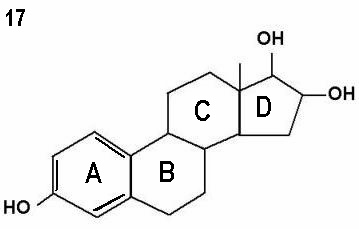
Figures 15 - 17 show oestrone, oestradiol and oestriol molecules showing one,
two and three hydroxyl groups respectively. Note the ketone group (=O)
attached to the D ring instead of a hydroxyl group in oestrone.
Oestrogen production and clearance are affected by the stage of the
menstrual cycle in premenopausal women. Furthermore, more than 95%
of the circulating oestradiol is produced by the ovary containing the
dominant follicle or corpus luteum (30). After the menopause, ovarian
oestrogen production and clearance decline substantially. Most of the
circulating oestradiol and oestrone production result from extra glandular
aromatisation of testosterone and androstenedione. Increased body fat
will increase such aromatisation, resulting in higher circulating levels of
oestradiol and oestrone (31).
Oestradiol is the main biologically active oestrogen in premenopausal
women. It circulates in the blood in 3 forms, bound to proteins,
conjugated in bile salts, and 2% as a free active fraction. About 60%
of oestradiol in circulation is bound to albumen, and 38% to SHBG. On
the other hand, oestrone is not strongly bound to plasma proteins,
with a higher metabolic clearance rate than oestradiol. At the same
time, oestrone forms the first step in the biological inactivation of oestradiol. This is followed by its conversion to oestrone sulphate,
catechol oestrogens, as well as oestriol and epioestriol (32). Catechol
oestrogens are so named because of their structural similarity to
catecholamines of having two hydroxyl groups on the aromatic A ring
(33). The main compounds in this subgroup are 2-hydroxyoestrone
and its metabolite 2-methyloestrone. The oestrogenic effects of
catechol oestrogens are limited to the central nervous system, but
have antioestrogenic effect in other oestrogen sensitive organs. All
metabolites are biologically less active than oestradiol itself (34).
Oestrogens function through genomic or non-genomic effects. The
genomic effect is imposed through their nuclear receptors, leading to
specific changes in gene transcription. As for progestogens and
androgens, the effects of oestrogens will be studied under 3 headings:
1. Chemical and endocrine;
Chemical and endocrine effects of oestrogens
Oestradiol is the main oestrogen in the non-pregnant young female. It is
mainly produced by the granulosa cells during the follicular phase and the
corpus luteum during the luteal phase. The rising levels of oestrogen
during the middle of the follicular phase reduce FSH production by the
pituitary gland through the negative feedback mechanism. This allows
mono-follicular growth, as the dominant follicle continues growing in
response to lower levels of FSH, unlike the smaller ones which stop
growing and become atretic. This is because the dominant follicle has
more FSH receptors, and higher aromatase enzyme activity which allow
easy conversion of androgens to oestradiol. It also has more LH receptors
and a rich micro vascular capillary network. Accumulation of androgens in
the smaller follicles is an important factor leading to their demise. The
ability of the dominant follicle to produce enough oestradiol to initiate the
negative feedback mechanism is important for mono-follicular ovulation.
This may not be the case in older women in their late 30s or early 40s, due
to the age related change in hypothalamic sensitivity. Accordingly, the
pituitary gland continues producing more FSH till 2 or 3 follicles are
capable of producing enough oestradiol to initiate the negative feedback
mechanism, and reduce FSH production. This is the scientific reason why
women in their late reproductive years are more likely to have
spontaneous multiple pregnancies.
Oestradiol is responsible for inducing hepatic production of SHBG,
thyroxine binding globulin and transcortin which are carrier molecules for
androgens and oestrogen, thyroxine, and cortisol respectively.
Accordingly, it has an important role to play in regulating the free
fractions of these hormones, and their metabolic clearance rate. Other
non-genomic effects of oestrogens include protein anabolic activity,
though to a lesser extent than that of androgens. They also have anti
osteoporotic effect as they promote bone deposition and reduce its
resorption. Oestrogen receptors have been isolated in bone. Oestradiol is
also known to cause vasodilatation, due to its direct activation of the
potassium channels in the plasma membranes. This leads to potassium
exit and relaxation of the vascular smooth muscle fibres. High doses of
oestradiol can cause excessive sodium and water retention, and lead to
high blood pressure.
Morphological effects of oestrogens
The main effects of oestradiol in this respect are:
-
Oestrogens in general have no direct or indirect role in the
development of female organs during intrauterine fetal life.
Nonetheless, maternal use of exogenous synthetic oestrogens
may lead to abnormality of the vagina and shape of the uterus, as
shown by the diethylstilbestrol effect. Offsprings of these mothers
developed vaginal polyposis, as well as T-shaped uterine
configuration;
-
Initiation of breasts development and its growth to adult size with
the help of other hormones including progesterone and prolactin;
-
Linear acceleration in height at puberty and final closure of the epiphyseal plates;
-
Cornification of the vaginal skin, and increase in total vaginal size;
-
Increasing the size of the cervix, uterus and tubes in preparation to reproductive function;
-
Deposition of fat in certain areas of the body which is a female physical characteristic;
-
Oestradiol increases endometrial cells hyperplasia and proliferation during the follicular phase. Together with progesterone they
sustain endometrial secretory changes and activity in preparation
for implantation during the luteal phase;
-
Oestradiol induces midcycle changes in the quantity and quality of
the cervical mucus to facilitate sperm migration into the upper uterine cavity. The cervix also acts as sperm reservoir to facilitate
continuous sperm availability for many hours after intercourse.
Natural non-human oestrogens
The most commonly used oestrogen in this group is premarin which is
a conjugated equine product. It is isolated from mares urine. The
composition of this product is made of: - 48% oestrone sulphate;
- 26% equilin sulphate which is the major circulating oestrogen in women using conjugated equine oestrogens. It is 4 times more
potent than the oestrone part, and is stored in the adipose
tissues;
- 15% 17α-dihydroequilin sulphate.
Premarin has been used extensively orally as hormone replacement
therapy mainly for vasomotor symptoms, and as vaginal cream for
postmenopausal vulvovaginal atrophic changes. Injectable forms are also
available, and the use of intravenous premarin in acute cases of severe
uterine bleeding has been mentioned before in this chapter.
Synthetic oestrogens These chemicals are divided into steroidal and non-steroidal compounds,
and have different functional characteristics in comparison to natural
oestrogens. The most commonly used steroidal ones are ethinyl oestradiol
and mestranol which is 3-methyl ether of ethinyl oestradiol. Examples of
the non-steroidal group include diethylstilbestrol, chlorotrianisene,
clomiphene citrate and tamoxifen. Few of these synthetic oestrogens have
very long nuclear retention. They act accordingly as oestrogens in a single
dose, but as oestrogen receptor modulators in repeated doses. This is
secondary to downregulation of the cytosol receptors, and inhibition of
messenger RNA transcription, due to prolonged nuclear occupation.
This effect can also be tissue specific, as these drugs act as anti
oestrogens in one tissue, and as oestrogens in others. A good example
is tamoxifen which has a very potent antioestrogenic effect on the
breast tissues, through its metabolite hydroxyl tamoxifen. A similar
antioestrogenic effect at the level of the hypothalamus is utilised for
induction of ovulation, by stimulating gonadotrophins secretion.
Conversely, it has an oestrogenic effect on the myometrium and
the endometrium, through different metabolites. This explains the endometrial hyperplasia, polyps and carcinoma risks reported with
prolonged use of tamoxifen by patients who had breast cancer.
Many drugs inhibit oestrogens synthesis directly by interfering with the
aromatase enzyme activity, without any direct effect on the cytoplasmic
receptors. The most commonly known ones in this group are letrozole
and anastrozole which are non-steroidal drugs. They can be used as
anti oestrogens for different purposes including induction of ovulation,
and for the treatment of endometriosis.
Oestrogens potency
Oestrogens potency depends on the time the oestrogen-receptor
complex occupies the nucleus of the target organ, after a single dose.
Oestrone and oestriol occupy the same receptors as oestradiol, but
have shorter nucleus retention time. Accordingly, they have weaker
biological effects than oestradiol, but repeated doses of either
hormone may have equivalent effects as a single oestradiol dose (35).
The nuclear retention time of oestradiol was found to be 1-4 hours.
Diethylstilbestrol had a longer time of 6-24 hours, and tamoxifen
retention time was 24-48 hours. Oestrogens potency has been tested
against the following parameters in postmenopausal women:
The ability to build up a proliferative endometrium; The ability to induce cornification of the vagina; The ability to reduce FSH and LH levels.
For these tests to be of any value in comparing different oestrogens,
many factors should be taken into consideration:
-
The type and dose of the oestrogen used;
-
The route of administration is very important. Conversion of oestradiol to oestrone takes place after oral administration by
the enzyme 17-keto reductase, which is available in the
gastrointestinal tract, but not in the vagina. Accordingly, vaginal
administration is more likely to increase oestradiol rather than
oestrone blood level. Similarly transdermal administration has
a similar effect, as oestradiol escapes the first pass through
the gastrointestinal tract and the liver, and its conversion to
oestrone;
-
The absorption rate and whether it is affected by other factors;
-
The metabolic clearance rate which depends on the blood level of
the carrier molecules, and hence the free fraction of the hormone;
-
The particular system under evaluation.
Use and side effects of oestrogen therapy
The most common uses for oestrogens in female patients in
chronological age order are: - To initiate pubertal development in cases of delayed puberty;
- In different contraceptives in combination with progestogens;
- For the treatment of dysfunctional uterine bleeding;
- In preparation of the endometrium during ovum donation cycles;
- Hormone replacement therapy in cases of surgical or natural menopause, and in cases of gonadal dysgenesis.
Side effects of synthetic oestrogens cover a wide range of organs and
effects. The most publicised risks following unopposed oestrogen use
are endometrial hyperplasia and carcinoma of the endometrium.
Accordingly, a progestogen should be used for a minimum duration of
12- 13 days with oestrogen HRT. This is not necessary in patients who
already had a hysterectomy. Further discussions about the relationship
between HRT and breast cancer, or cardiovascular disease will be found
in Chapter 9. Other side effects of synthetic oestrogens include: Hypertension is a risk in susceptible patients, due to increased
plasma renin activity and renin substrate. There is also increased
aldosterone production and sodium retention.
There is a two-foldincreasedthromboembolictendency,causedby
increased production of factors II, VII, X and fibrinogen. This is
coupled with decreased antithrombin III activity. The final outcome
is a hypercoagulable state. This risk is especially high in heavy
smokers, diabetics, and with previous history of thrombosis.
Certain conditions may also increase this risk including
immobilisation, trauma or surgical procedures, sepsis and obesity.
The route of administration also has an important effect. There is
more thrombosis risk with the oral route than the transdermal or
vaginal routes, because of the first hepatic pass of oestrogens, and
increased production of coagulation factors with the oral route.
Ischaemic heart disease risk is also increased because of the
increase in triglycerides level, despite the favourable effects of
oestrogens on HDL cholesterol and LDL cholesterol.
Cholelithiasis is also a side effect of synthetic oestrogens. The
relative risk in postmenopausal women is 2.5, compared to those
who are not using HRT. This effect may follow:
a. Alteration in lipid balance; b. Alteration in bile salts content; c. Alteration in HDL cholesterol.
Contraindication to oestrogen use
Taking into account all the metabolic, endocrine and anatomical points
mentioned before, oestrogens should not be used in the following
conditions:
With undiagnosed genital bleeding; In case of acute liver disease; Present or past history of oestrogen dependent cancer; History of thromboembolism.
Few conditions should be taken into consideration in a risk / benefit
assessment, before prescribing oestrogens. These conditions include:
History of liver disease; Diabetes mellitus; Uterine fibroids; Familial porphyria cutanea tarda.
Antioestrogens
Antioestrogens are substances that compete with oestrogens at their
binding receptor sites. This effect can be universal, or specific to few but
not all tissues. Clomiphene citrate and tamoxifen are the classical
examples in this group. Tamoxifen acts as an antioestrogen at the
breasts, but stimulates oestrogen receptors in the uterus, which may
result in hyperplasia and polyps, or even endometrial carcinoma. The
role of catechol oestrogens as antioestrogens outside the central
nervous system (CNS) has been mentioned before. Within the CNS,
they compete with catecholamines for the enzyme catechol-
methyltransferase, which results in reduced degradation of CNS
catecholamines. This will prolong the effects of catecholamines
within the brain, with consequent modulation of catecholamines
sensitive hypothalamic releasing and inhibiting factors (32). Though
progestogens are usually considered to have an antioestrogenic effect,
they tend to exhaust rather than occupy oestrogen receptors. So
strictly speaking progestogens modify oestrogen effects, but do not
compete with them for their receptor sites.
Summary
It is evident that progestogens, androgens, and oestrogens have similar
basic steroidal rings, yet subtle changes in those molecules gave them different endocrine, morphological and metabolic characteristics. Such
features may even be different within the different subgroups of each
hormone. This depends on the chemical structure, half-life and
bioavailability, affinity to receptors, potency and metabolic clearance
rate. More clinical information will be provided in Chapters, 9, 10, 12 and
13. It is also important to take the information provided in this chapter
into consideration, when reading the chapters dealing with hormone
replacement therapy and contraception.
- Knobil E and Hotchkiss J. The menstrual cycle and its
neuroendocrine control. In: Knobil E, Neill J. Eds. The Physiology of Reproduction. New York: Raven Press, 1994; 711 - 749.
-
Blank SK, McCartney CR and Marshall JC. The origins and
sequelae of abnormal neuroendocrine function in polycystic ovary
syndrome. Hum Reprod 2006; 12(4): 351 361.
-
Zarutskiea PW and Phillips JA. Reanalysis of vaginal progesterone
as luteal phase support (LPS) in assisted reproduction (ART)
cycles. Fertil Steril 2007; 88 (supplement 1): S113.
-
Schust DJ, Anderson DJ, and Hill JA. Progesterone induced
immunosuppression is not mediated through the progesterone
receptor. Hum Reprod 1996; 11(5): 980 985.
-
Landau RL and Lugibihl K. The effect of progesterone on the
concentration of plasma amino acids in man. Metabolism 1967;
16(12): 1114 1122. 6. Kalkhoff RK. Metabolic effects of progesterone. Am J Obstet Gynecol
1982; 142(6 Pt 2): 735 738. 7. Sitruk-Ware R. Pharmacological profile of progestogens. Maturitas
2009; 47(4): 277 - 283. 8. Städtler F, Langner V. The effect of cyproterone and gonadotrophins
on the adrenal gland of juvenile and adult rats. A morphological and
morphometrical study. Pathol Res Pract 1985; 179 (4-5): 493 498.
-
Honer C, Nam K, Fink C, Marshall P, Ksander G, Chatelain R,
Cornell W, Steele R, Schweitzer R, Schumacher C. Glucocorticoid
receptor antagonism by cyproterone acetate and RU486′′. Mol
Pharmacol 2003; 63 (5): 1012 1020
-
Pham-Huu-Trung M, de Smitter N, Bogyo A, Girard F. Effects of
cyproterone acetate on adrenal steroidogenesis in vitro. Horm Res
1984; 20 (2): 108 115.
-
Johansson EDB. Depression of the progesterone levels in women
treated with synthetic gestagens after ovulation. Acta Endocrinol
1971; 68(4): 779 792.
-
Stevenson JC. Hormone replacement therapy and lipids.
Menopause Review 1997; 2: 1520.
-
Crook D, Cust MP, Gangar KF, Worthington M, Hillard TC,
Stevenson JC, Whitehead MI, Wynn V. Comparison of transdermal
and oral oestrogen-progestin replacement therapy: effects on
serum lipids and lipoproteins. Am J Obstet Gynecol. 1992;
166(3): 950 - 905.
-
Crook D, Godsland IF, Hull J, Stevenson JC. Hormone replacement
therapy with dydrogesterone and 17 beta-oestradiol: effects on
serum lipoproteins and glucose tolerance during 24 month follow
up. Br J Obstet Gynaecol. 1997; 104(3): 298 - 304.
-
Depoprovera Product Monograph. Depoprovera. Pfizer, Canada
Inc, 2006.
-
Preston JT, Cameron IT, Adams EJ and Smith SK. Comparative study
of tranexamic acid and northisterone in the treatment of ovulatory
menorrhagia. Br J Obstet Gynaecol 1995; 102: 401 - 406.
-
Irvine GA, Campbell-Brown MB, Lumsden MA, Heikkilä A, Walker
JJ, Cameron IT Randomised comparative trial of the
levonorgestrel intrauterine system and northisterone for
treatment of idiopathic menorrhagia. Br J Obstet Gynaecol 1998;
105: 592 - 598.
-
Andersson JK, Rybo G. Levonorgestrel-releasing intrauterine
device in the treatment of menorrhagia. Br J Obstet Gynaecol.
1990; 97(8): 690 - 694.
-
Vercellini P, Viganò P, Somigliana E The role of the levonorgestrel-
releasing intrauterine device in the management of symptomatic
endometriosis. Curr Opin Obstet Gynecol. 2005; 17(4): 359 - 365.
-
Bergeron C. Morphological changes and protein secretion induced by
progesterone in the endometrium during the luteal phase in
preparation for nidation. Hum Reprod 2000; 15 (Suppl 1): 119 - 128.
-
Vereide AAB, Kaino T, Sager G, Orbo A. Scottish Gynaecological
Clinical Trial Group, Bcl-2, BAX, and apoptosis in endometrial
hyperplasia after high dose gestagen therapy: a comparison of
response in patients treated with intrauterine levonorgestrel and
systemic medroxyprogesterone. Gynecol Oncol 2005; 97: 740 - 750.
-
Hedderson MM, Ferrara A, Williams MA, Holt VL and Weiss NS.
Androgenicity of progestins in hormonal contraceptives and the
risk of gestational diabetes mellitus. Diabetes Care 2007; 30(5):
1062 1068.
-
Swyer GIM. Determination of progestational potency: a review.
J Roy Soc Med 1984; 77: 406 409.
-
Greenblatt RB, Jungck EC and Barfield WE. Anew test for
efficiency of progestational compounds. Ann N Y Acad Sci 1958;
71(5): 717 - 721.
-
Longcope C. Adrenal and gonadal androgen secretion in normal
females. Clin Endocrinol Metab1986; 15: 213 228.
-
Longcope C, Sato k, McKay C and Horton R. Aromatization by
splanchnic tissue in men. J. Clin Endocrinol Metab 1984; 58: 1089
1093.
-
Ishimaru T, Edmiston WA, Pages L and Horton R. Splanchnic
extraction and conversion of testosterone and dihydrotestosterone
in man. J Clin Endocrinol Metab 1978; 46: 528 533.
-
Walters KA, Allan, and Handelsman DJ. Androgen actions and the
ovary. Biol Reprod 2008; 78: 380 - 389.
-
Aziz R, Carmina E and Sawaya ME. Idiopathic hirsutism. Endocr
Rev 2000; 21: 347 362.
-
Baird DT and Frase IS. Blood production and ovarian secretion
rates of oestradiol-17/3 and oestrone in women throughout the
menstrual cycle. J Clin Endocrinol Metab 1974; 38: 1009 1017.
-
Jud HL. Hormonal dynamics associated with the menopause; Clin
Obstet Gynecol 1976; 19(4): 775 788.
-
Ruder HJ, Loriaux L and Lipsett MB. Oestrone sulphate:
Production rate and metabolism in man. J Clin Invest 1972; 52:
1020 1033.
-
Fishman J. The catechol oestrogens. Neuroendocrinology 1976;
22(4): 363 374.
-
Buster J. Oestrogen kinetics for clinicians. Glob libr womens med.
ISSN: (1756 2228) 2008; DOI 10.3843/GLOWM 10280.
-
Sasson S and Notides AC. Oestriol and oestrone interaction with
the oestrogen receptor. J Biol Chem 1983; 258(13): 8113 - 8117
|
|